Overview of CoeViz 2
About
CoeViz 2 is a web-based tool for the analysis and visualization of covarying positions (residues) in proteins. For a given protein sequence, the web-server computes pairwise coevolution scores using either of the three metrics: Chi-square statistic, Pearson correlation, or Mutual Information. In contrast to the previous version (CoeViz), CoeViz 2 represents covarying amino acids as graph nodes and focuses on the identification of cliques. Large scale analysis on 7595 non-redundant proteins suggests that such cliques frequently represent functional residues involved in the same function.
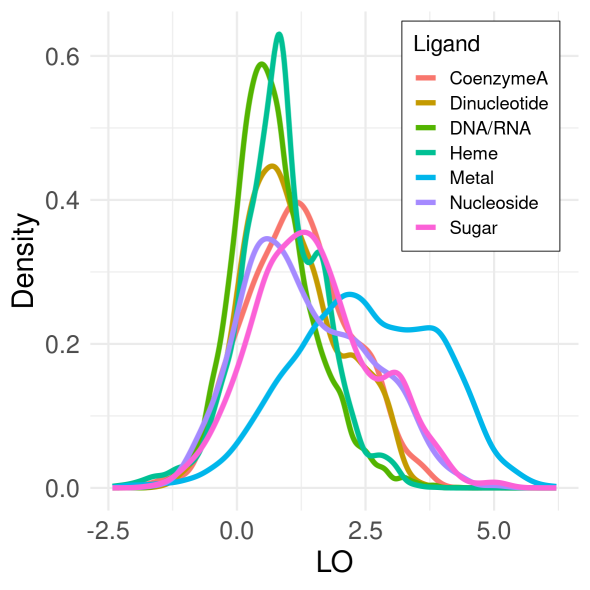
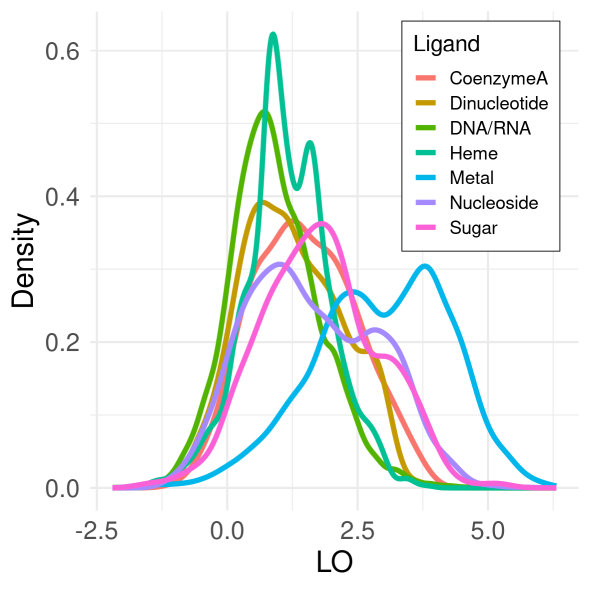
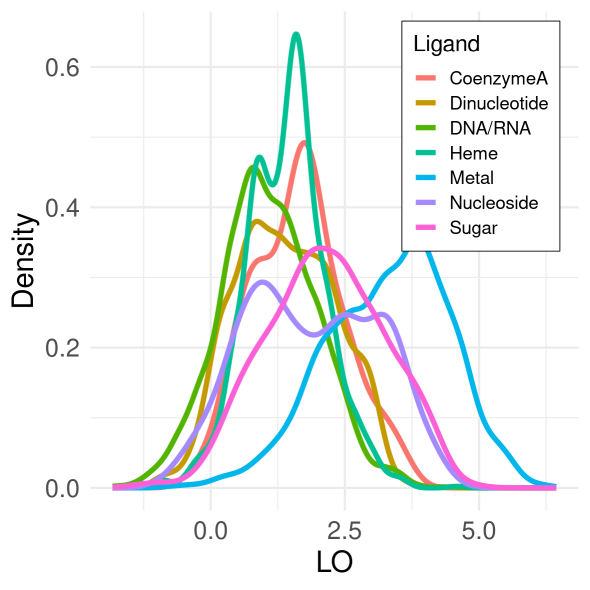
Figure 1. Density distributions of normalized scores (LO) for covarying residues that bind the same natural ligand. Here, distribution computed over 7595 non-redundant proteins using Chi-squared covariance metric after 0.3, 0.4, or 0.5 cutoff applied.
Terms of use and disclaimer
All data and images generated by the CoeViz 2 server can be freely saved,
printed, and distributed by means of any media without our written
permission for academic and non-commercial purposes. However, the use
of CoeViz 2 results should be acknowledged by a reference to the server.
The use of the CoeViz 2 server is at your own risk and no
liability is accepted for any loss or damage arising through the use
of the web site and protein annotations generated by the server.
Reference to cite the server
To cite CoeViz 2, please use the following reference:
Corcoran D, Maltbie N, Sudalairaj S, Baker FN, Hirschfeld J, Porollo A.
CoeViz 2: Protein Graphs Derived From Amino Acid Covariance.
Front. Bioinform. 2021 Jun 24;1:653681. doi: 10.3389/fbinf.2021.653681
Methods
At the base of CoeViz 2 are the same covariance metrics used as in CoeViz, see their formulations here. However, they are all computed based on multiple sequence alignments adjusted for phylogenetic bias only. All other alternative adjustment schemes not provided here for clarity, as they did not improve the identification of covarying functional residues.
In addition to the 2-letter alphabet (amino acid at a given position in the query sequence versus any other amino acid), CoeViz 2 now has an option of computing the same covariance metrics using the 20-letter alphabet (probabilities computed over all 20 natural amino acids). Though the latter option appears not as informative for the identification of functional residues, it could be useful for comparison with other covariance methods.
CoeViz 2 accepts as input a protein sequence of any length, including multi-domain proteins. The largest protein tested (using the 2-letter alphabet for generating a covariance matrix) was about 7000 amino acids long. Although the longer sequence is, the more resources (memory and CPU time) will be required to render such protein as a graph in a web browser.
Enrichment of the covariance neighborhood for functional residues was computed as follows. For the residue at position i, residues (r) among the all covarying positions (n) with covariance scores cov(i,j) ≥ given cutoff (c) involved in the same MF (f) represent the nearest neighbor covariance environment and are quantified via the normalized score LO:
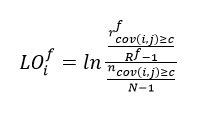
where Rf is the total number of residues with the MF f in a given protein, N is the total number of residues in the protein; both adjusted for the residue of interest.
User Manual
The main input expected from the user is a protein sequence that needs to be specified in the FASTA format. Additional options include a covariance metric, amino acid alphabet (2 or 20) to compute positions specific probabilities, and the source of multiple sequence alignment, either based on UniProt UniRef90, Pfam, or a user provided. The default values for these additional options are chosen as suboptimal based on the evaluation of 7595 proteins.
The resulting page of CoeViz 2 consists of two parts, a navigation panel on a left hand side and an interactive protein graph view area. At the navigation panel, user can change a covariance cutoff to define graph cliques, review the precomputed distribution stats per different cutoffs, and download generated data. At the interactive view area, the user can move the graph, drag the nodes, zoom-in/out the view, and review specific values for nodes and edges. For the original CoeViz visualization using interactive heatmaps, hierarchical trees, circular diagrams, and multi-dimensional scaling 3D plots (MDS), one can use a CoeViz link at the bottom of the navigation panel.
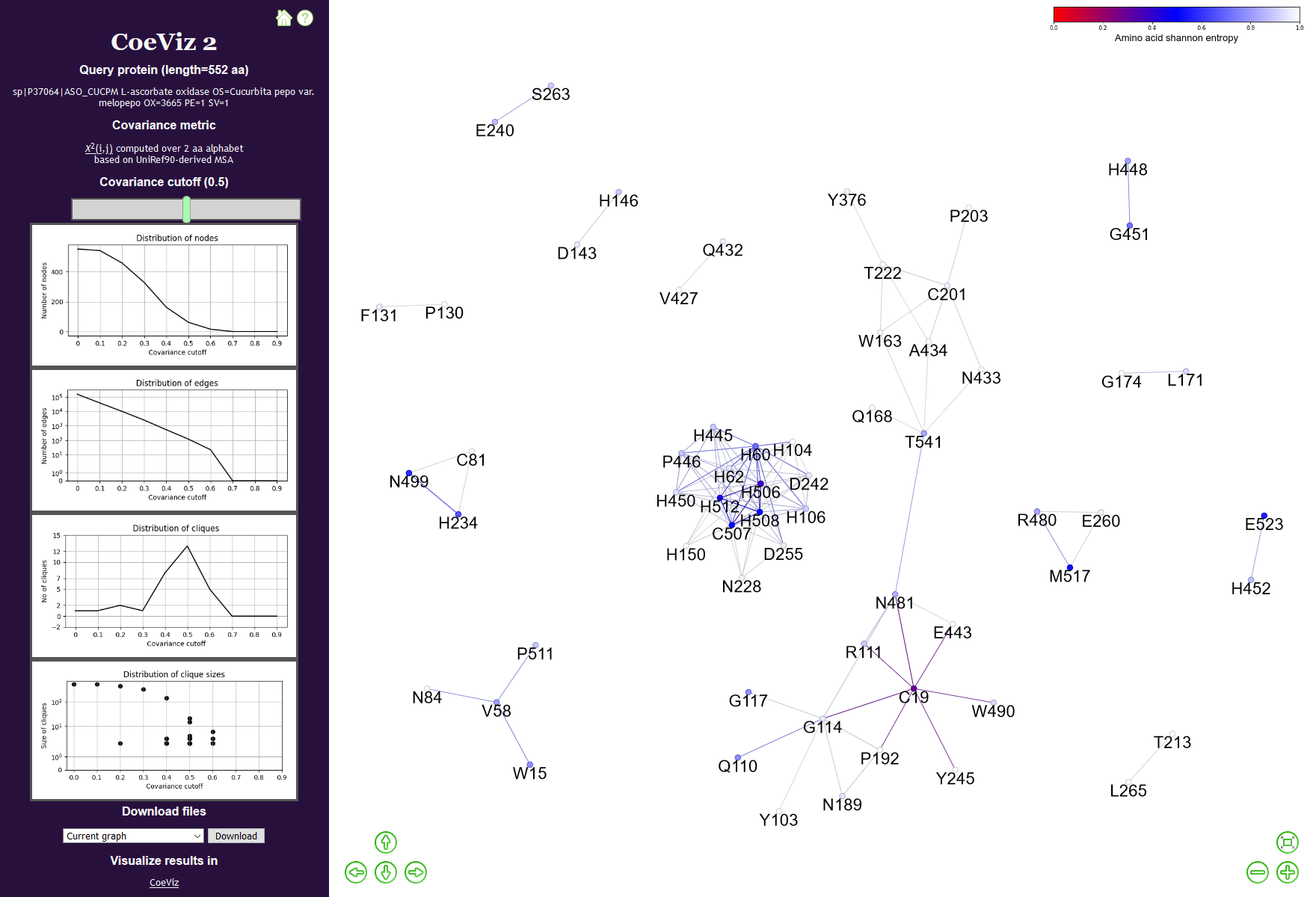
Figure 2. The resulting page of CoeViz 2. The user can alter a covariance cutoff to change the number of nodes and cliques to view. Distributions of various protein graph parameters are precomputed and provided below the slider to guide the user on results per a given cutoff. At the view area, the user can drag the entire graph or individual nodes, zoom-in on a specific clique, and mouse hover of graph elements to review specific data: amino acid residue, its conservation score, pairwise covariance data, and corresponding values from the inverse matrix.
Try the example presented at Figure 2 interactively.