About
This website is updated as new data and tools become available.
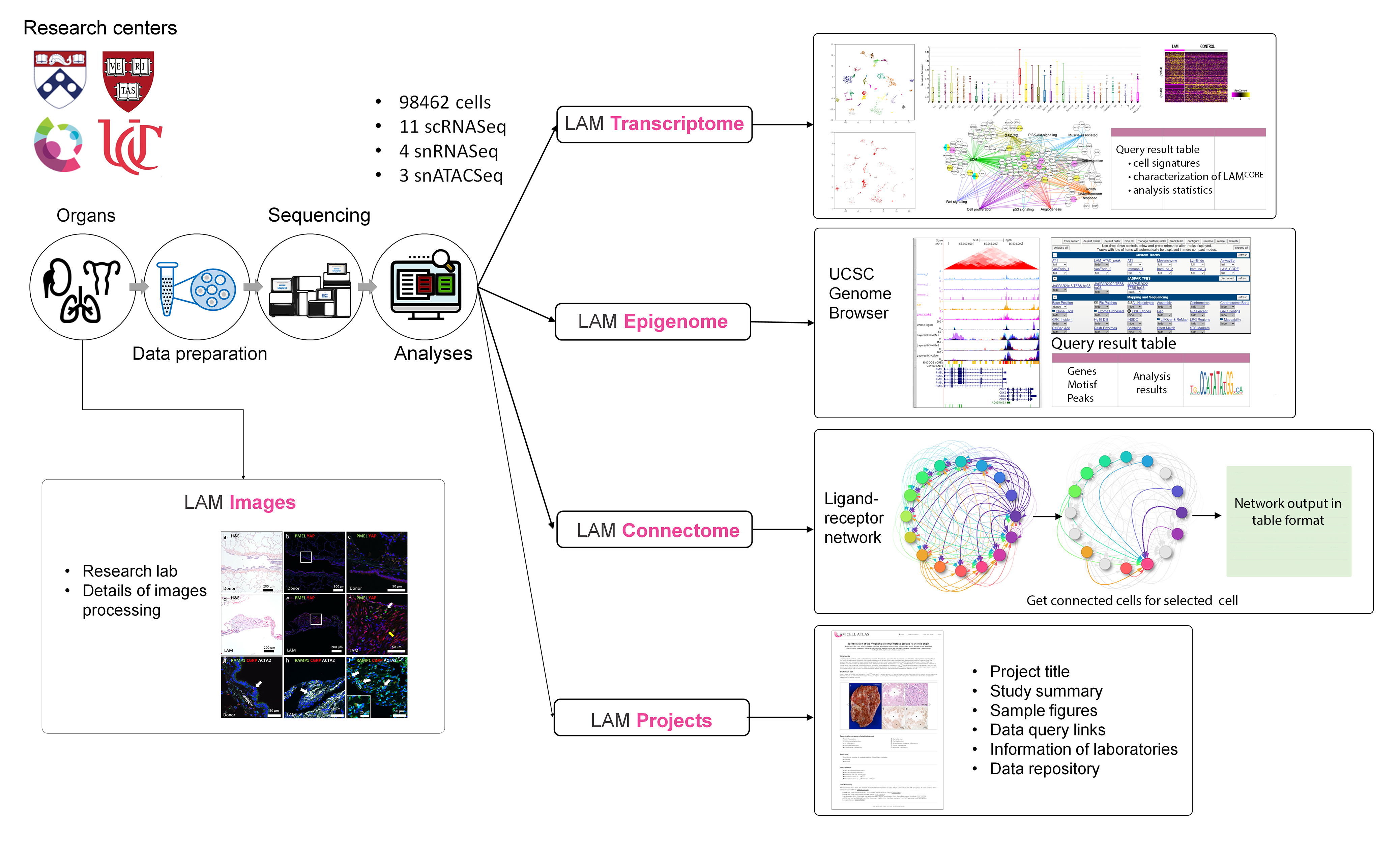
LAM Transcriptome
Integrative analysis of the single cell and single nucleus RNA-seq data from LAM lungs (n=13) and the uterus (n=1) from a patient with pulmonary LAM, and a LAM patient with a kidney angiomyolipoma (AML, n=7) identified heterogeneous cell types and selective signature genes for each predicted cell type. "LAM Transcriptome" provides the query functions including "Gene query", "Cell type query" and "Characterization of LAMCORE Cell".
Query by gene
"Query by gene" allows users to input any gene symbol of interest. The expression patterns and scRNA-seq analytic statistics of the queried gene are visualized in UMAP (Uniform Manifold Approximation and Projection), Box, Notched Box, Beeswarm, Scatter plot, and bi-directional bar chart. In addition, LAM Cell Atlas provides internal links to LungMAP datasets hosted in the LGEA (Lung Gene Expression Analysis) database and provides external links by mapping query gene to external knowledgebase and resources such as NCBI, GeneCard, MGI, Ensembl, UniPort and The Human Protein Atlas. By linking these internal and external databases, users can readily incorporate gene expression profiles across multiple lung developmental time points and disease conditions to gain a more comprehensive understanding of the gene of interest.
Cell type query
"Cell type query" enables users to select one of the predefined cell types within LAM lung, uterus or renal AML to query the database. For each cell type of interest, the LCA provides cell selective gene signatures. Featured signature genes are displayed in a searchable and sortable table along with an interactive bar graph which enables users to compare the mean gene expression across all cell types. We previously performed single cell transcriptome analysis and identified a unique population of cells termed LAMCORE that are distinct from, but closely related to, lung mesenchymal cells. The pulmonary LAMCORE shared genetic, morphological and transcriptomic similarity to uterine LAMCORE and co-expressed multiple known LAM markers such as ACTA2, PMEL, FIGF (VEGF-D), and MLANA. In the present study we further validated the presence of lung LAMCORE in separate and/or integrated datasets of LAM lung samples from independent data collections and different single cell platforms.
Characterization of LAMCORE
"Characterization of LAMCORE" web query functions are designed to enable users to access the LAMCORE signature (genes selectively expressed in LAMCORE cells in comparison with all other cells in the LAM lung), the LAMCORE secretome (LAMCORE signature genes encoding proteins predicted to be secreted proteins) and the differentially expressed genes in LAMCORE cells vs. lung mesenchymal cells from control female lungs. Taken together, the query functions in "LAM transcriptome" are designed to facilitate the retrieval of cell-specific gene expression data across all cells with a special focus on LAMCORE cells based on previous publications and ongoing integrative analysis.
LAM Epigenome
"LAM Epigenome" is designed to query genes and display chromatin accessibility of different LAM lung cells based on snATAC-seq analyses. The "Gene Query" function displays queried genes using an embedded UCSC Genome Browser (https://genome.ucsc.edu/) with enabling of selected LAM snATAC-seq data tracks. Users can retrieve chromatin accessibility signals at the queried gene locus across all LAM cell-types. Tracks of DNase hypersensitivity and signal density of histone marks (H3K4Me1, H3K4Me3, and H3K27Ac) in multiple cell types can be visualized using overlapping-colored graphs. The "Cell Type Query" function allows users to explore cell specific motifs identified through our snATAC-seq analyses. Corresponding transcription factors associated with the enriched motifs are coded using hyperlink format. By this method, users can quickly browse peak signals of LAM snATAC-seq data and search for enriched motifs associated with the cell type of interest in the UCSC Genome Browser.
LAM Connectome
Since only a small fraction (<20%) of the cells in LAM lesions contain TSC gene mutations,8 it is likely that most cells in the LAM nodules are recruited by the LAMCORE cells via cell-cell communication. To understand the complex interactions of LAMCORE cells with host stromal cells that influence LAM progression and metastasis, we designed "LAM Connectome" based on the ligand-receptor interaction analysis using Connectome (https://github.com/msraredon/Connectome) and the FANTOM5 ligandreceptor database. Users can select the ligand-receptor family of interest from the drop-down list and obtain the given ligand-receptor pair interactions across different LAM lung cell types. Outputs are visualized as weighted, directed connectome networks as well as an interactive table. For each specific signaling network, users can identify the corresponding source and receiver cells of the ligand-receptor pair with ranks based on the weighted value for two connected cells (i.e., the product of z-score transformed mean expression of ligand and receptor pairs). For example, when "VEGF" family is selected from the drop-down list, the top three ranked ligand-receptors are "VEGFD-FLT4", "VEGFD-NPR2" and "VEGFD-ITGA9" with LAMCORE cells as the source cell and lymphatic endothelial cells (LymEndo) as the receiver cells. The network plot is interactive to enable users to click on a cell of interest to highlight the connected nodes and edges and gray-out the other unconnected elements in the network. Users can also visualize the interaction edges by clicking on the edge to inspect the specific ligand-receptor pairs. Other web interactive features including move, zoom-in and out, and download are also included for optimal navigation. The "LAM Connectome" allows researchers to explore cellular signaling and cell-cell communication in LAM lung cells and predicted ligand-receptor pairs for further hypothesis driven functional interrogation and experimental validation.
LAM Projects
The "LAM Projects" was designed as a storyboard to introduce single-cell omics applications related to LAM. In addition to introducing the publication (publication title, author list, abstract, and figures) and contributing laboratories, LCA provides a dataset query function (to access interpreted date via gene or cell query), data repository links (to access raw data from GEO or dbGAP), and GitHub (to access the analytic pipeline used in the manuscript) if those components are available in the published manuscript. Current "LAM Projects" include recent publications from Guo et al. 2020, Obraztsova et al. 2020, Tang et al. and Al Mahi et al. Using this storyboard, we can introduce the single cell applications in LAM around the world and LAM researchers can browse and access the fully processed single cell omics data to inform their own research directions.
LAM Network
(Under Development for Version 2 release)
Gene Regulatory Networks (GRNs) represent the complex interactions between transcription factors (TFs) and their target genes, controlling cell fate and cell state transitions at both healthy and diseases conditions. To reveal key regulators driving LAM pathogenesis, we constructed LAMCORE cell specific GRN using PECA2, a statistical model designed to infer gene regulatory networks from paired gene expression (RNA-seq) and chromatin accessibility (ATAC-seq) data. Through the integrative single cell multiomic data analysis, we reported the activation of a uterine-specific HOX-PBX gene regulatory network that regulates the survival of LAM cells.(PMID: 37163604)
LAM ST
(Under Development for Version 2 release)
While single-cell transcriptomics are powerful to reveal cellular heterogeneity, identify and characterize novel cell types or cell states, and understand cellular responses to diseases, it does not identify regional gene expression changes associated with diseases processes. Spatial transcriptomics (ST) has rapidly emerged as a groundbreaking technology, enabling researchers to map gene expression within the native tissue architecture. By preserving the spatial context of cellular interactions, ST provides unprecedented potential to reveal insights into tissue and organ architecture, cell-cell communication, and disease mechanisms. Current visium and xenium LAM spatial transcriptome data is contributed by Drs . Minzhe Guo, Jane Yu and Yan Xu and supported by LAM Foundation (LAM0150C01-22)
LAM ShinyCell
(Under Development for Version 2 release)
ShinyCell provides a user-friendly interface for researchers to explore processed LAM single-cell datasets without the need for coding expertise. Through interactive UMAP/t-SNE visualizations, customizable gene expression plots, metadata filtering, and downloadable outputs, ShinyCell allows users to rapidly navigate cellular landscapes and interrogate genes or pathways of interest.
Contact
References
1. Du Y, Guo M, Wu Y, Wagner A, Perl AK, Wikenheiser-Brokamp K, Yu J, Gupta N, Kopras E, Krymskaya V, Obraztsova K, Tang Y, Kwiatkowski D, Henske EP, McCormack F, Xu Y. Lymphangioleiomyomatosis (LAM) Cell Atlas. Thorax 2023;78(1):85-87. PMID:36599466.
2. Guo M, Yu JJ, Perl AK, Wikenheiser-Brokamp KA, Riccetti M, Zhang EY, Sudha P, Adam M, Potter A, Kopras EJ, Giannikou K, Potter SS, Sherman S, Hammes SR, Kwiatkowski DJ, Whitsett JA, McCormack FX, Xu Y. Single-Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell.Am J Respir Crit Care Med 2020;202(10):1373-1387. PMID:32603599.
3. Obraztsova K, Basil MC, Rue R, Sivakumar A, Lin SM, Mukhitov AR, Gritsiuta AI, Evans JF, Kopp M, Katzen J, Robichaud A, Atochina-Vasserman EN, Li S, Carl J, Babu A, Morley MP, Cantu E, Beers MF, Frank DB, Morrisey EE, Krymskaya VP. mTORC1 activation in lung mesenchyme drives sex- and age-dependent pulmonary structure and function decline.Nat Commun 2020;11(1):5640. PMID:33159078.
4. Tang Y, Kwiatkowski DJ, Henske EP. Midkine expression by stem-like tumor cells drives persistence to mTOR inhibition and an immune-suppressive microenvironment.Nat Commun 2022;13(1):5018. PMID:36028490.