Integration of transcriptomic and proteomic data identifies biological functions in cell populations from human infant lung
Abstract
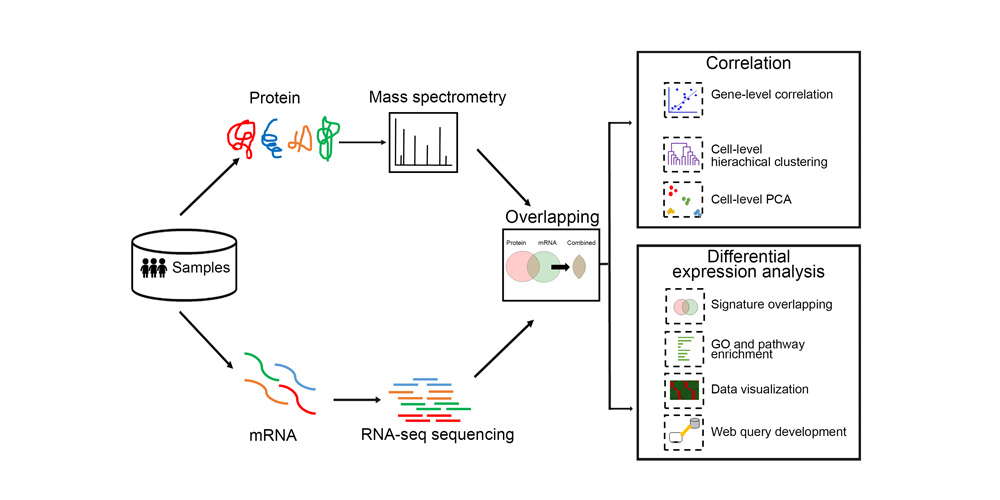
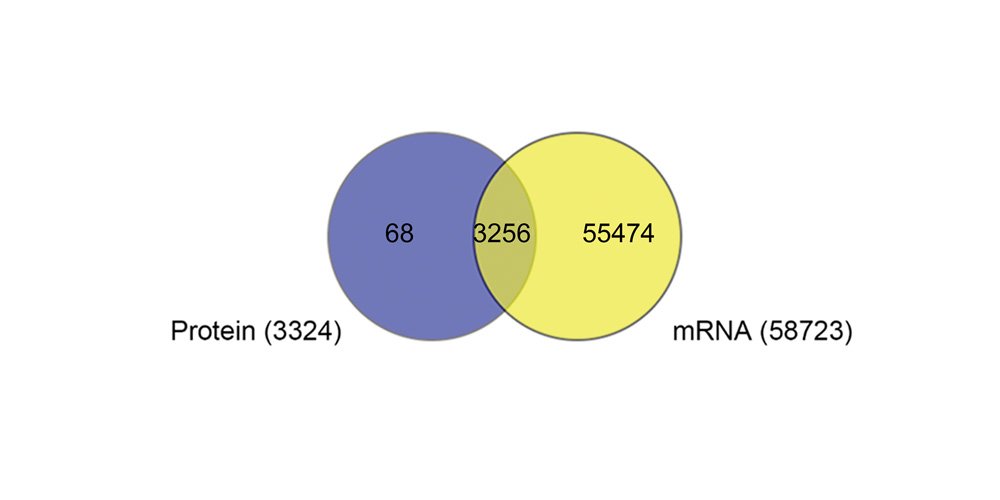
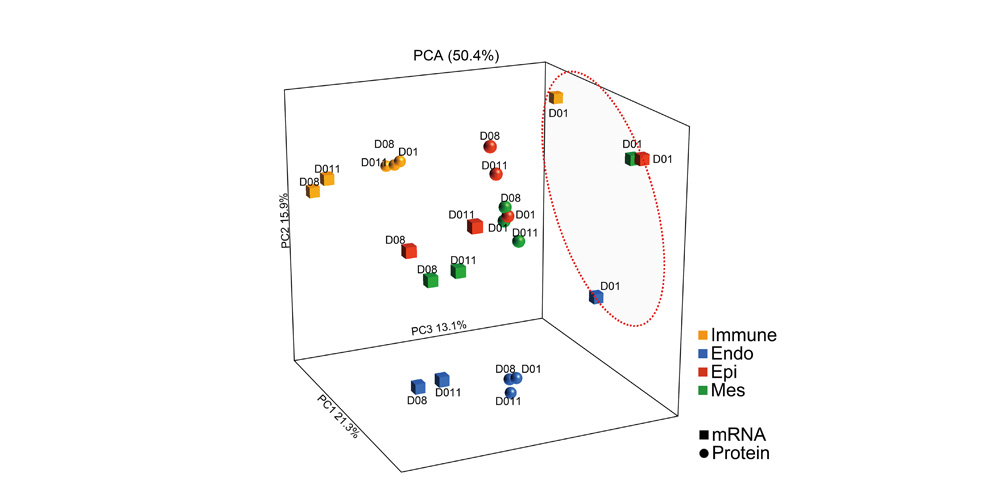
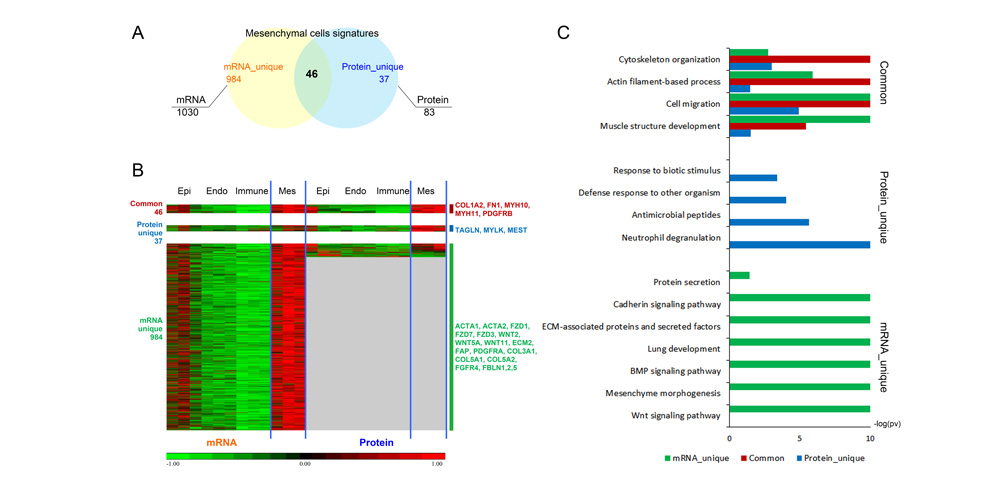
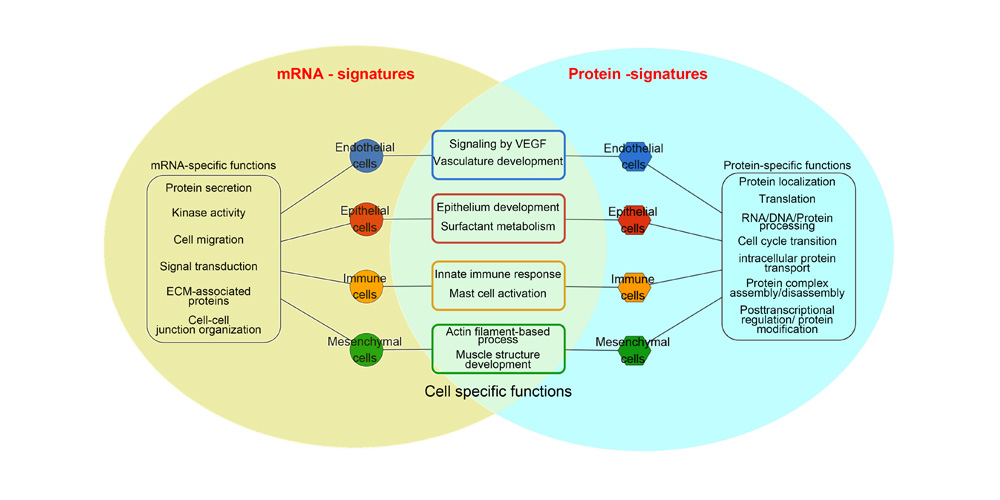
Systems biology uses computational approaches to integrate diverse data types to understand cell and organ behavior. Data derived from complementary technologies, for example transcriptomic and proteomic analyses, are providing new insights into development and disease. We compared mRNA and protein profiles from purified endothelial, epithelial, immune, and mesenchymal cells from normal human lung tissue. Signatures for each cell type were identified and compared at both mRNA and protein levels. Cell specific biological processes and pathways were predicted by analysis of concordant and discordant RNA-protein pairs. Cell clustering and gene set enrichment comparisons identified shared versus unique processes associated with transcriptomic and/or proteomic data. Clear cell-cell correlations between mRNA and protein data were obtained from each cell type. Approximately 40% of RNA-protein pairs were coherently expressed. While the correlation between RNA and their protein products was relatively low (approximately 0.4), cell specific signature genes involved in functional processes characteristic of each cell type, were more highly correlated with their protein products. Consistency of cell specific RNA-protein signatures indicated an essential framework for the function of each cell type. Visualization and reutilization of the protein and RNA profiles are supported by a new web application, "LungProteomics," which is incorporated into the LGEA web portal and freely accessible at (https://research.cchmc.org/pbge/lunggens/lungProteomics/human_sorted_profiles.html).
Query functions
Download
Links




· LGEA Web Portal @ CCHMC 2014-2018 · ALL RIGHTS RESERVED ·