Sincera:
A Computational Pipeline for Single Cell RNA-Seq Profiling Analysis
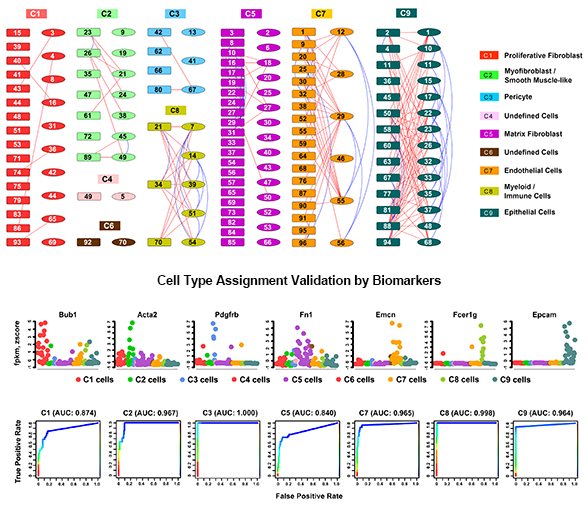
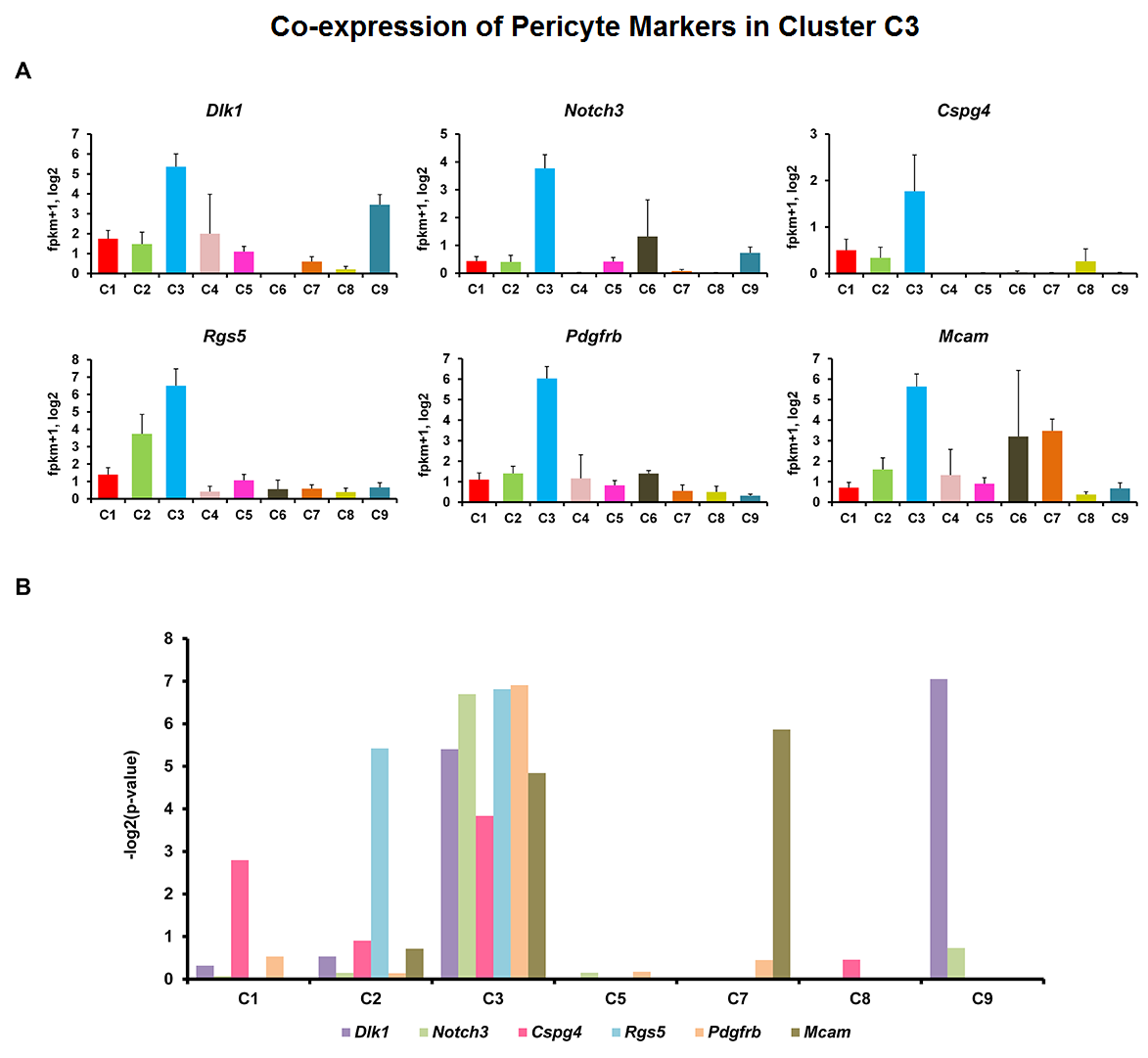
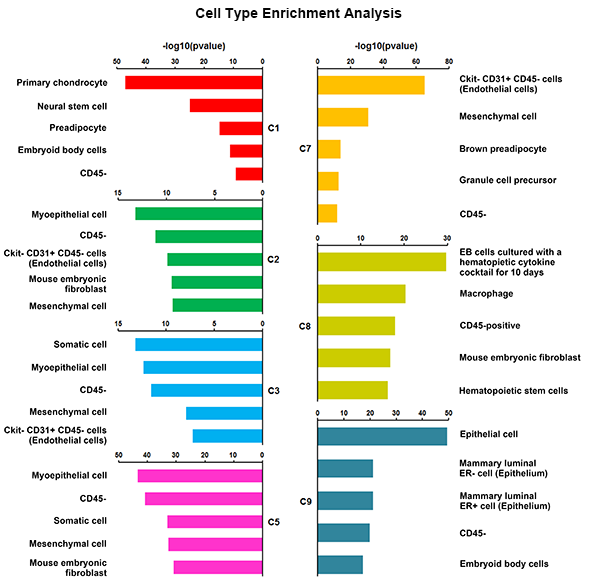
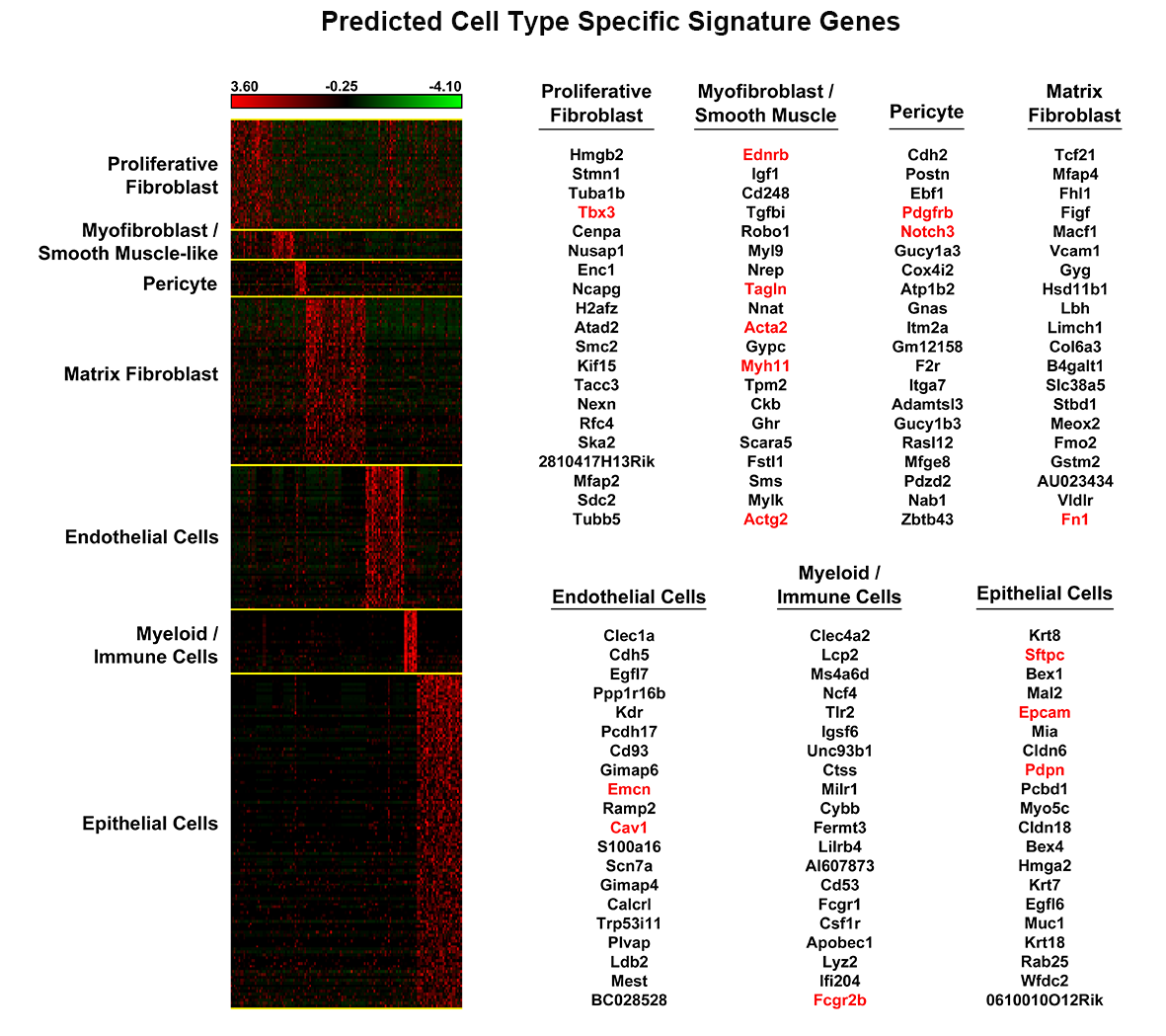
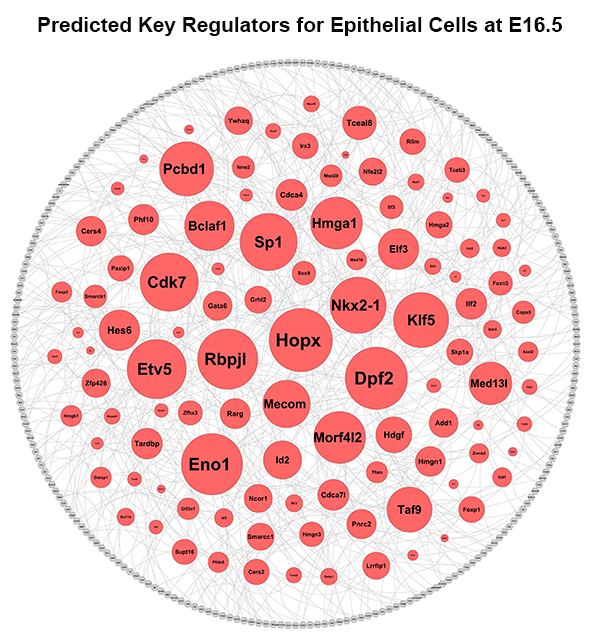
We applied this pipeline to RNA-seq analysis of single cells isolated from embryonic mouse lung at E16.5. Through the pipeline analysis, we can distinct major cell types of fetal mouse lung, including epithelial, endothelial, smooth muscle, pericyte, and fibroblast-like cell types; identify gene signatures, bioprocesses, and key regulators associated with each cell type.
• Minzhe Guo, Hui Wang, S. Steve Potter, Jeffrey A. Whitsett, Yan Xu. SINCERA: A Pipeline for Single-Cell RNA-Seq Profiling Analysis. PLoS Comput Biol. 2015 Nov 24;11(11):e1004575. doi: 10.1371/journal.pcbi.1004575. eCollection 2015.
SINCERA is now on github
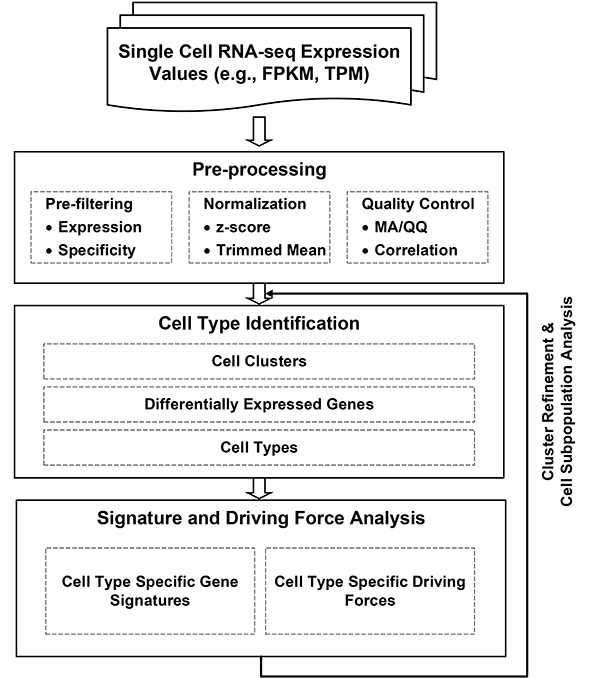
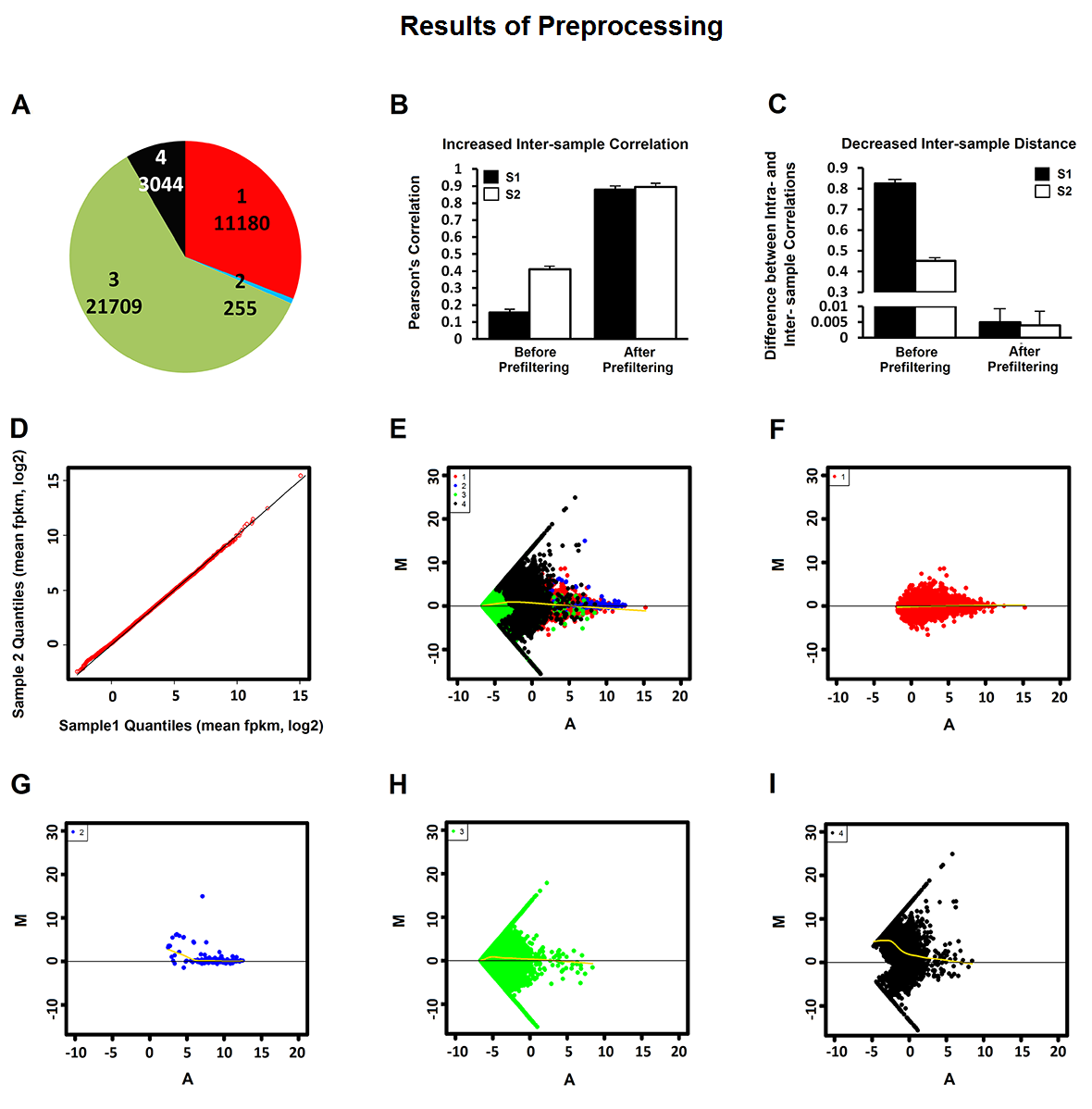
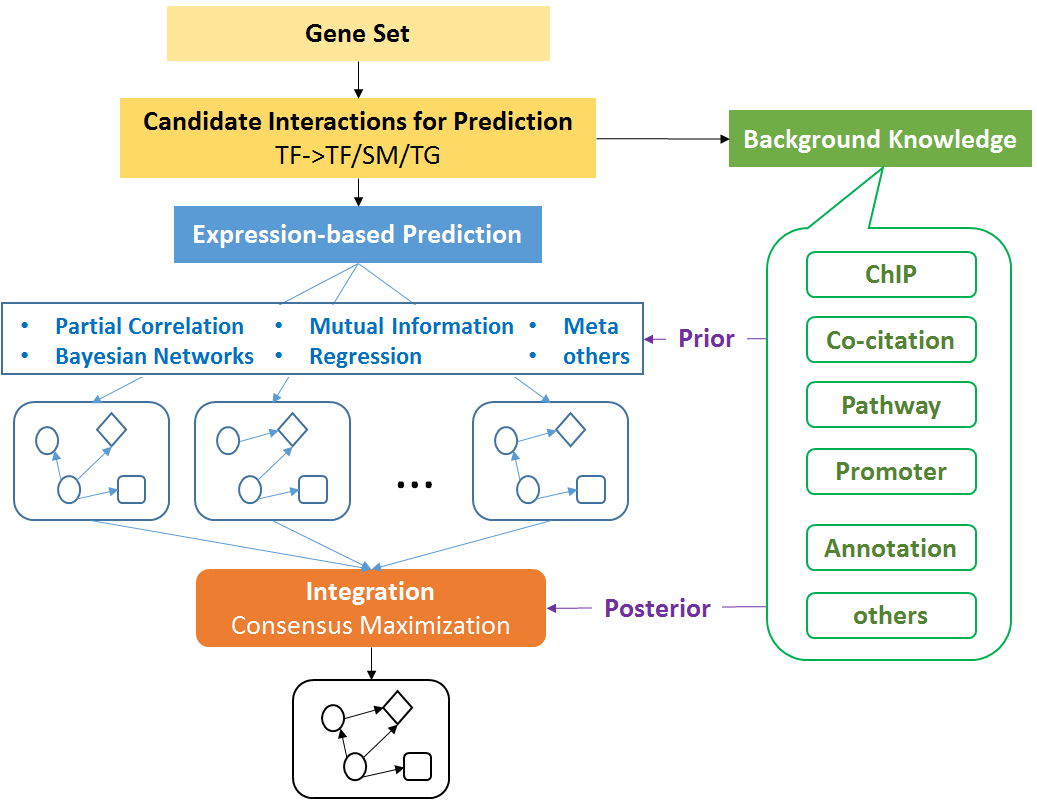
The pipeline takes RNA-seq expression values (FPKM, TPM, or raw counts) from heterogeneous single cell populations as inputs. It consists of three major components: preprocessing, cell type identification, cell type specific gene signature and driving force analysis. The output of the pipeline includes a set of cell clusters, differentially expressed genes for each cluster, the gene signatures and driving forces of a given cell cluster. Each cluster is considered as a unique cell type with defined biological functionality.
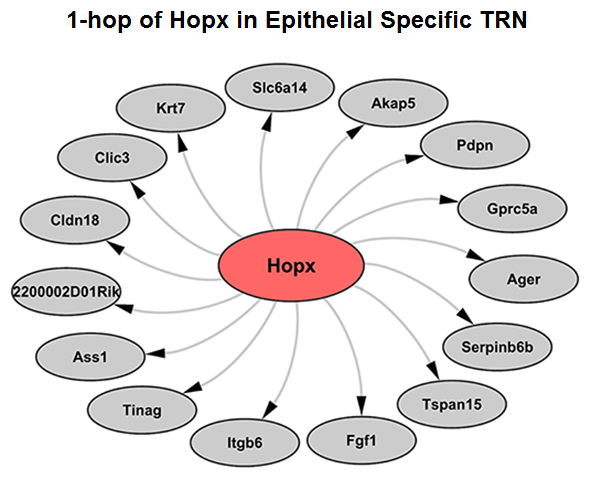
Considering the heterogeneity of cell states at a given developmental stage, sub-clusters are likely present in each major cluster. The procedures of cell type identification, gene signature prediction, and driving force analysis can be iterated and refined to subpopulations of cells.
The pipeline is designed to enable researchers analyzing their single cell transcriptome data on their desktop or laptop. The initial sequencing data mapping, alignment, quantification and annotation can be processed using software such as Tophat, BWA, Cufflinks, and RSEM, which are not covered in this pipeline.